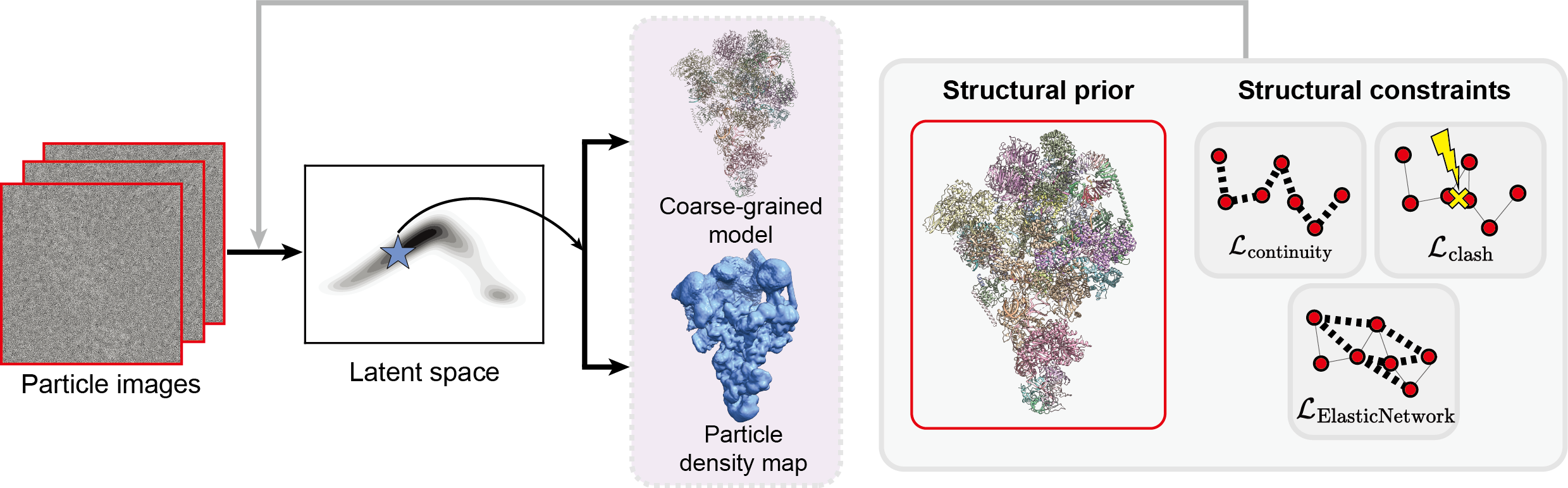
Resolving conformational heterogeneity in cryo-electron microscopy (cryo-EM) datasets remains a significant challenge in structural biology. Previous methods have often been restricted to working exclusively on volumetric densities, neglecting the potential of incorporating any pre-existing structural knowledge as prior or constraints. In this paper, we present a novel methodology, cryoSTAR, that harnesses atomic model information as structural regularization to elucidate such heterogeneity. Our method uniquely outputs both coarse-grained models and density maps, showcasing the molecular conformational changes at different levels. Validated against four diverse experimental datasets, spanning large complexes, a membrane protein, and a small single-chain protein, our results consistently demonstrate an efficient and effective solution to conformational heterogeneity with minimal human bias. By integrating atomic model insights with cryo-EM data, cryoSTAR represents a meaningful step forward, paving the way for a deeper understanding of dynamic biological processes.


@article{li2023cryostar,
author={Li, Yilai and Zhou, Yi and Yuan, Jing and Ye, Fei and Gu, Quanquan},
title={CryoSTAR: leveraging structural priors and constraints for cryo-EM heterogeneous reconstruction},
journal={Nature Methods},
year={2024},
month={Oct},
day={29},
issn={1548-7105},
doi={10.1038/s41592-024-02486-1},
url={https://doi.org/10.1038/s41592-024-02486-1}
}